
|
BY: QUEST You may be wondering what a “molecular simulation” is anyway. We got this for you! Molecular simulations or molecular modeling is a collection of ways for doing “computer experiments” on model systems. Model systems are simplified version of real world cases. Molecular models are guided by the theory and work through the tools of math and physics. But then the scientists end up with equations that are too difficult to solve in a simple and accurate form. Not surprisingly, computers or supercomputers are used for numerical solutions. As you figured by now, this is a very interdisciplinary field and is called molecular modeling or computational chemistry/biology.  In the simplest terms, results from computer experiments or simulations are to be compared with the experimental data for validation. After that, they are used to make new predictions about natural phenomena. This saves us a lot of human time with no safety issues as often is the case in “wet lab” experiments. Molecular simulations have limitations and will probably never be able to replace wet lab experiments but can serve as a direct supplement to them. Therefore, the two are used in a complementary way to shed light on a wide range of natural phenomena. Today, we focus on a specific type of simulation called Molecular Dynamics (MD). MD simply simulates the time-dependent motion of molecules based on Newtonian mechanics. For biomolecules, you can imagine it like a biological microscope with a resolution of an angstrom (1x10^-10 meters!). Since a typical chemical bond length is about 1 angstrom, MD simulations can show how all atoms move: A real-time biological movie! 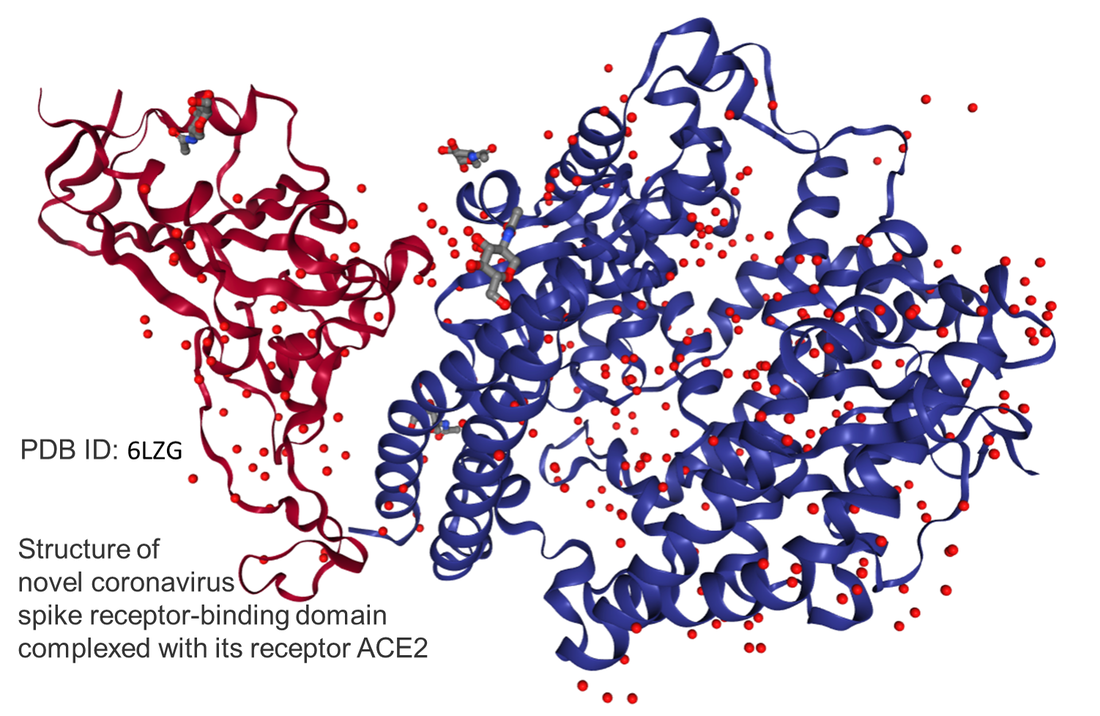 Since the beginning of the outbreak, the experimentalists have been working hard to reveal as much structural information as possible related to SARS-CoV-2 (the virus that causes COVID-19). By the way, here is a good summary regarding how to name this human coronavirus. Anyway, yes, we have lots of structural information already available in the protein databank. However, they are static (snapshot-like) 3D structures. Although they provide a lot of useful information, to find the druggable sites or understand the mechanism of entry to the body, motion of the virus proteins will be invaluable. In the past, MD simulations captured motions that create potentially druggable sites in numerous proteins including the Ebola protein. Also, keep in mind that the experimental structure itself is a must information for MD simulations to start with. Therefore, the experiment and computation go hand in hand!
We hope this gives you some insights regarding the molecular simulations. Drop a comment below to continue the discussion!
0 Comments
Leave a Reply. |
Quest Student Research InstituteOn Science, Computation, Medicine, and Academic Success Archives
January 2022
|
 RSS Feed
RSS Feed